
DRUGONE
变异的不完全显现性(即携带致病变异但未表现出相应疾病表型)是遗传变异解释中的重大挑战。研究人员利用 Genome Aggregation Database (gnomAD) 的80多万份人类基因组数据,对ClinVar数据库中已知的临床相关变异进行了系统分析,揭示了影响显现性的多种分子机制。研究人员特别分析了77个与高外显率、早发、严重单倍剂量不足疾病相关的基因中的734个预测性功能缺失变异(pLoF),发现其中95%的“未显现疾病”个体可由注释误差、体细胞变异、拼接修复或其他救援机制解释。真正无法解释的病例极为罕见,显示出深入的逐例分析对于减少误判疾病风险的重要性。

预测无症状个体的疾病风险是精准医学的核心目标之一。然而,许多个体虽携带致病变异,却并未表现出疾病表型,形成了所谓的不完全显现性。这一现象可能源于变异注释不准、环境与调控差异或其他修饰因子。以往的显现性研究往往基于症状明确的病患群体,容易因抽样偏差而高估风险。而大规模群体数据库,如UK Biobank或gnomAD,为更公正地评估显现性提供了新途径。
ClinVar汇总了来自临床与研究实验室的变异数据,但由于提交模式分散且更新不一致,其中部分“致病”标注可能存在误差。gnomAD作为全球最大的人群变异数据库,在其最新版本(v4)中包含约80万人基因组和外显子组数据,涵盖多样的种群背景,是研究变异显现性的重要资源。研究人员利用该数据库探讨致病变异为何能在健康个体中“被容忍”,并系统评估其潜在机制。
方法概述
研究人员整合ClinVar中约231万条变异数据,与gnomAD v4的807,162名个体的变异信息进行比对。首先,评估ClinVar变异在gnomAD中的覆盖率及不同版本间的变化;其次,重点分析预测性功能缺失变异(pLoF),通过逐例人工评估与功能注释规则体系(包含32条判定标准),识别导致假阳性或显现性降低的潜在机制,如转录本选择错误、低表达区、拼接修复、体细胞来源或技术伪影。最后,结合sQTL(剪接数量性状位点)分析,探索非编码调控如何通过可变剪接影响显现性。
结果
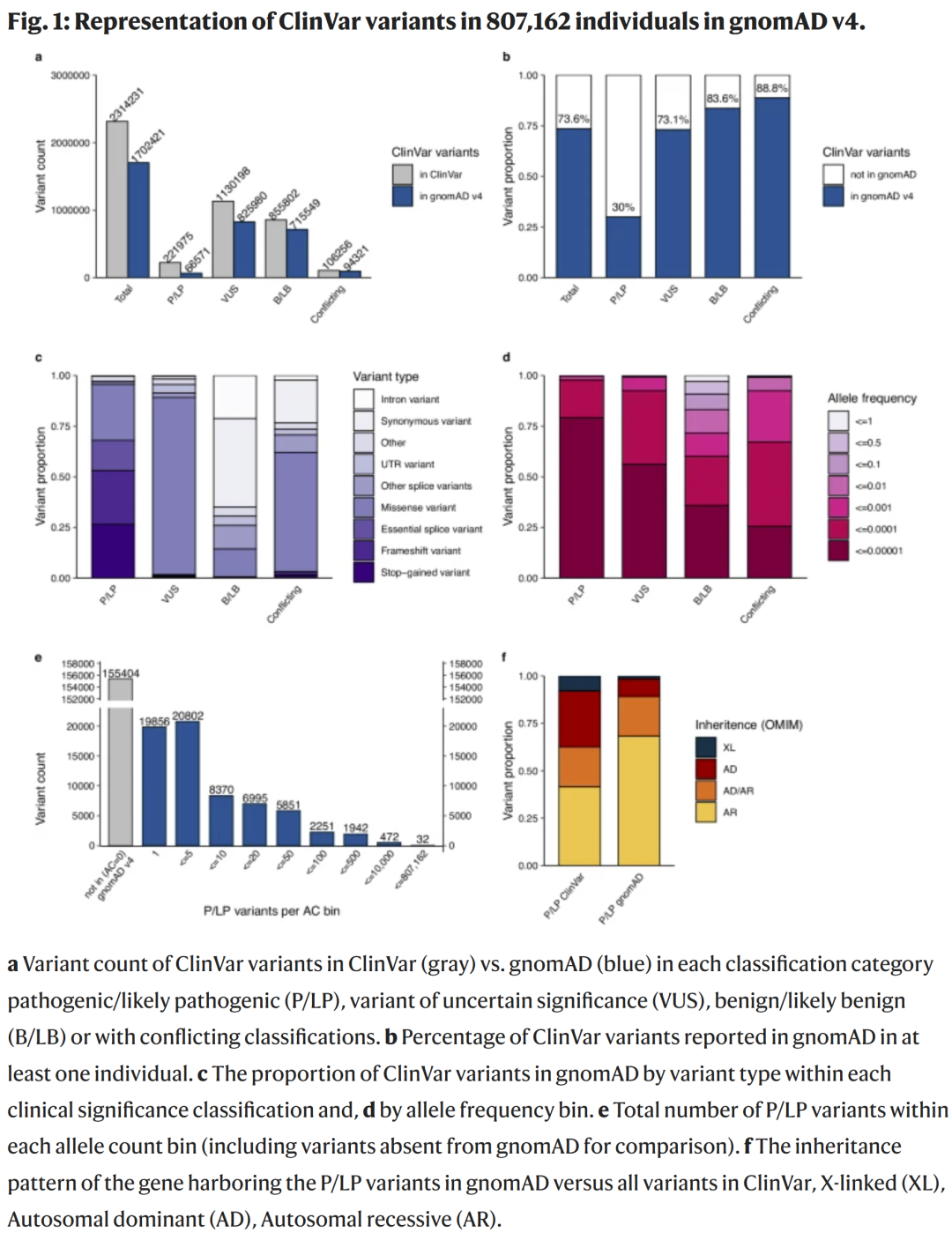
ClinVar变异在gnomAD中的覆盖显著提升
在231万条ClinVar变异中,约74%(1,702,421条)出现在gnomAD样本中。其中致病/可能致病(P/LP)变异的代表性为30%,不确定意义变异(VUS)为73%,良性/可能良性(B/LB)变异为84%。P/LP变异主要包括无义、移码及关键剪接位点变异,且绝大多数极为罕见(97.6%的等位基因频率 < 0.0001)。从gnomAD v2到v4,个体数增长约5.7倍,使ClinVar变异覆盖率从56.9%提升至73.6%,其中P/LP变异代表性几乎翻倍(从16.3%到30%)。这些变化说明更大规模的数据集显著提高了临床变异的表征精度。
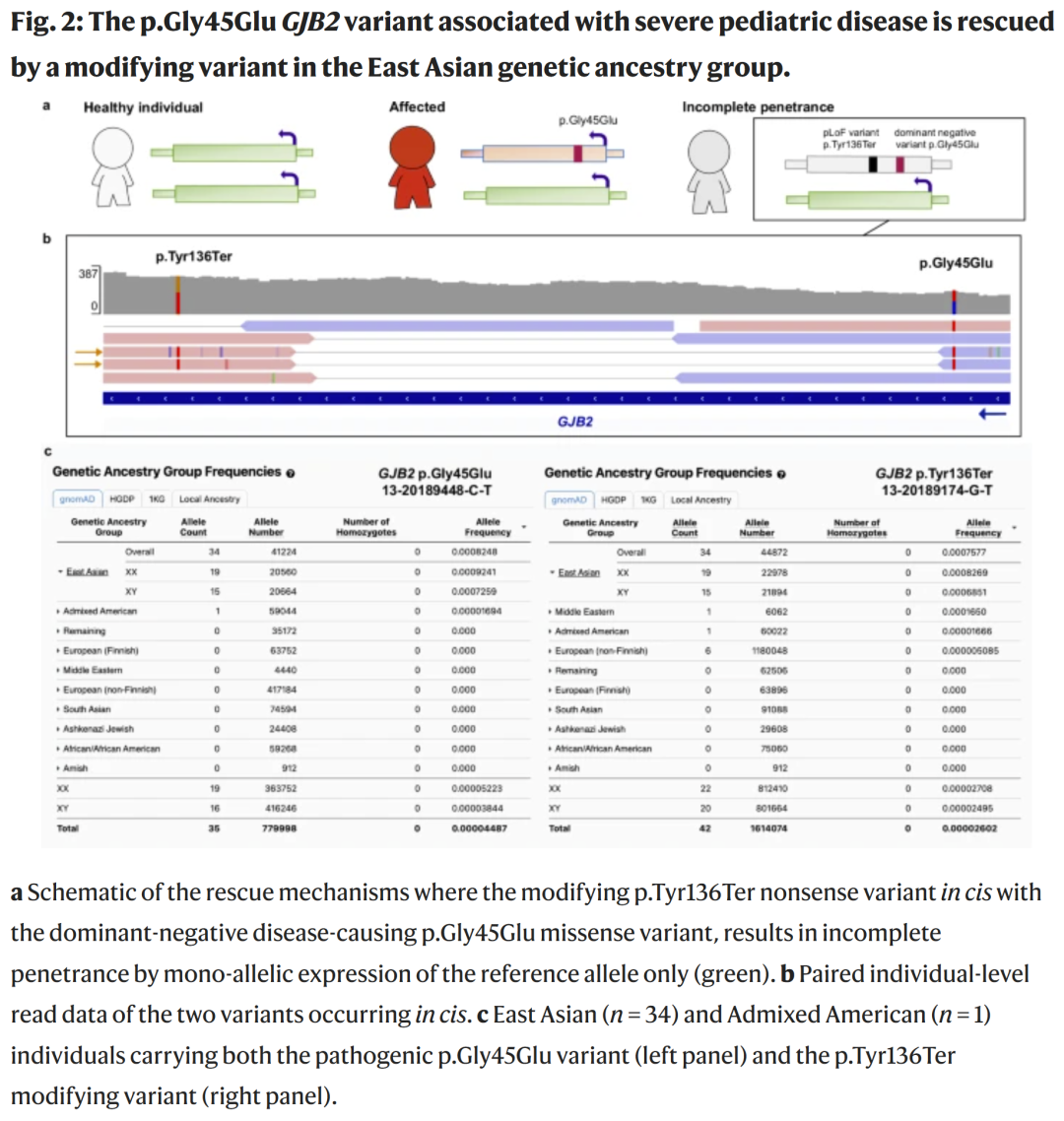
发现人群特异的不完全显现性实例
研究人员发现一个典型的群体特异性显现性调节案例:在东亚人群中,致病性 GJB2 p.Gly45Glu 变异(导致角膜炎-鱼鳞癣-耳聋综合征)在35名个体中出现,但所有人同时携带 p.Tyr136Ter 无义变异,两者位于同一单倍型上,后者“抵消”了前者的显性负效应,使这些个体未表现出致死性表型。这是一个罕见但清晰的“功能缺失救援”实例,展示了遗传背景在显现性调控中的关键作用。
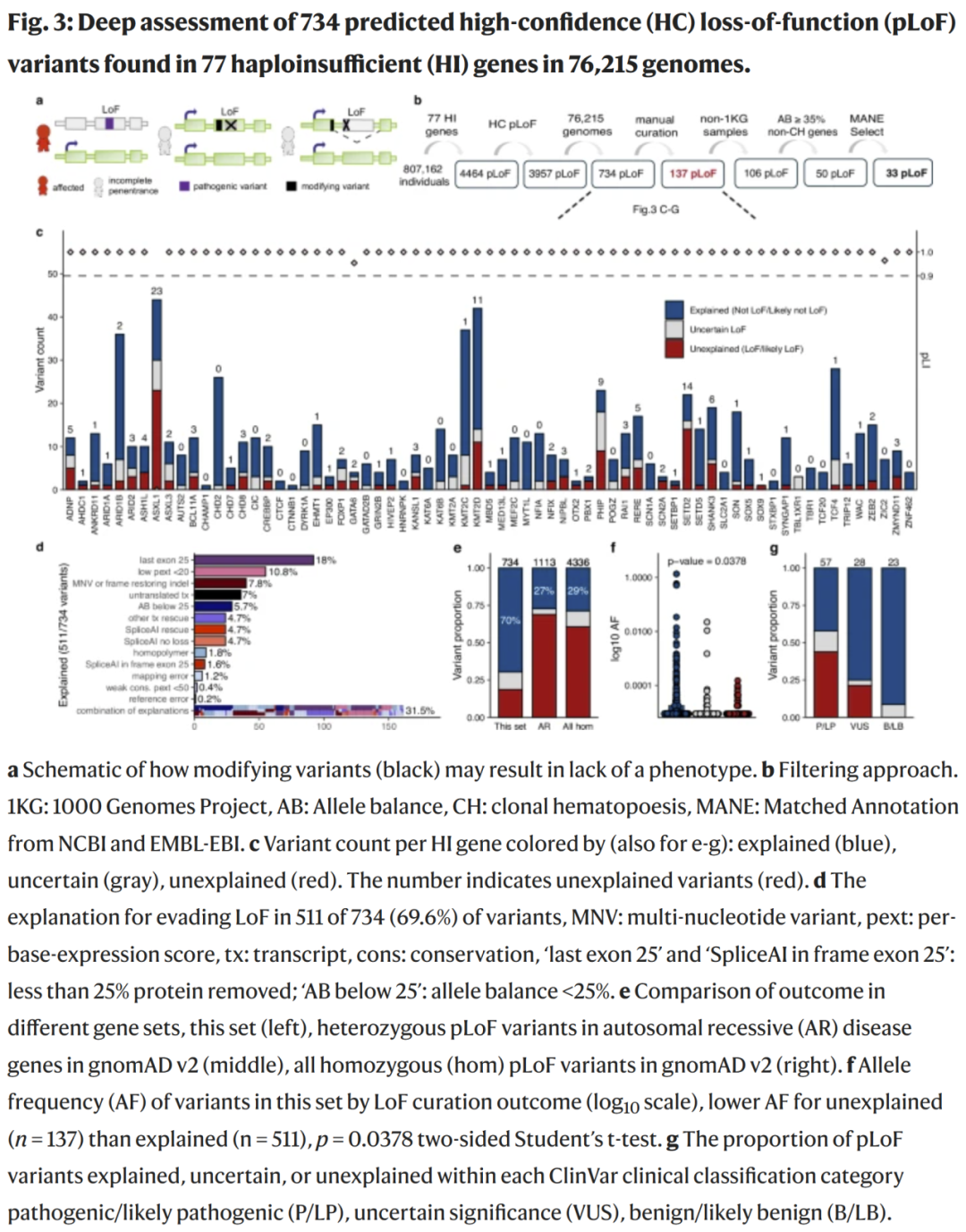
对77个高显现疾病基因的pLoF系统评估
在807,162个体中,共检测到4,464个pLoF变异,其中734个位于全基因组数据中、分布于77个高显现疾病相关基因。研究人员逐例分析发现,约70%的变异可被解释为并不真正导致功能丧失,主要原因包括:
位于终末外显子或其前50个碱基区域(18%),不触发无义介导的mRNA降解;
位于低表达区(10.8%);
被邻近多核苷酸变异或移码修复所“救援”(7.8%);
存在替代表达转录本(7%);
可能为体细胞事件(等位基因平衡 <25%,约5.7%)。
此外,11.7%的变异证据不足被归为“不确定”,仅18.7%的变异仍无法解释。这意味着绝大多数被标记为“高风险”的pLoF变异其实并不真正致病。
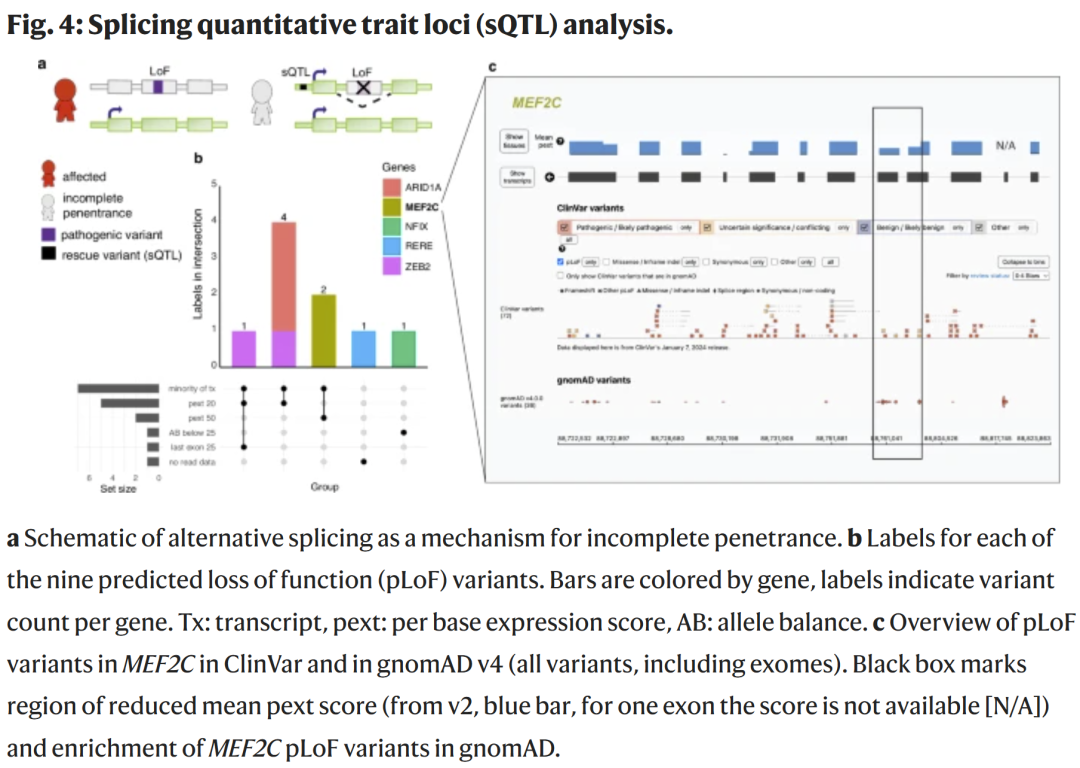
非编码sQTL介导的可变剪接导致的不完全显现
在734个pLoF变异中,有9个位点与特定的剪接数量性状变异(sQTL)共定位,提示其所在区域可能通过可变剪接调节显现性。以MEF2C基因为例,两个pLoF变异位于低表达区(pext值下降约50%),且携带sQTL的个体呈现外显子排除现象。MEF2C的功能缺失与神经发育障碍相关,这表明部分携带者因sQTL导致的剪接调节而未发病。此结果提示:sQTL及可变剪接是导致显现性降低的重要机制。
讨论
研究人员通过对80多万人基因组的系统分析,揭示了在健康个体中出现“致病变异”的多种原因。显现性降低的主因并非生物学异常,而往往源于注释错误、技术假阳性或体细胞突变。深入的逐例评估显著降低了“假致病”判定率,使变异解读更可靠。
此外,研究强调了多样人群的重要性。东亚人群中的GJB2案例表明,种群特异的单倍型结构可能显著影响显现性。研究还指出,样本来源(如细胞系或老年个体)会增加体细胞变异的干扰,因此在利用群体数据库时需谨慎。
通过LoF功能注释框架的系统化分析,研究人员得出结论:
约95%的pLoF变异“未致病”现象可被合理解释;
真正无法解释的不完全显现仅占4.5%;
sQTL介导的剪接调控是少数但确凿的显现性调节机制;
对每一个pLoF变异进行个体化评估是减少误判、改进临床遗传学的重要途径。
本研究不仅深化了对显现性和变异解释的理解,也为精准医学中的风险评估和遗传咨询提供了坚实的数据基础。
整理 | DrugOne团队
参考资料
Gudmundsson, S., Singer-Berk, M., Stenton, S.L. et al. Exploring penetrance of clinically relevant variants in over 800,000 humans from the Genome Aggregation Database. Nat Commun 16, 9623 (2025).
https://doi.org/10.1038/s41467-025-61698-x

内容为【DrugOne】公众号原创|转载请注明来源
内容中包含的图片若涉及版权问题,请及时与我们联系删除
